An aberrant STAT pathway is central to COVID-19 getmixapp
An aberrant STAT pathway is central to COVID-19 getmixapp
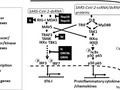
COVID-19 is caused by SARS-CoV-2 infection and characterized by diverse clinical symptoms. Type I interferon (IFN-I) production is impaired and severe cases lead to ARDS and widespread coagulopathy. We propose that COVID-19 pathophysiology is initiated by SARS-CoV-2 gene products, the NSP1 and ORF6 proteins, leading to a catastrophic cascade of failures. These viral components induce signal transducer and activator of transcription 1 (STAT1) dysfunction and compensatory hyperactivation of STAT3. In SARS-CoV-2-infected cells, a positive feedback loop established between STAT3 and plasminogen activator inhibitor-1 (PAI-1) may lead to an escalating cycle of activation in common with the interdependent signaling networks affected in COVID-19. Specifically, PAI-1 upregulation leads to coagulopathy characterized by intravascular thrombi. Overproduced PAI-1 binds to TLR4 on macrophages, inducing the secretion of proinflammatory cytokines and chemokines. The recruitment and subsequent activation of innate immune cells within an infected lung drives the destruction of lung architecture, which leads to the infection of regional endothelial cells and produces a hypoxic environment that further stimulates PAI-1 production. Acute lung injury also activates EGFR and leads to the phosphorylation of STAT3. COVID-19 patients’ autopsies frequently exhibit diffuse alveolar damage (DAD) and increased hyaluronan (HA) production which also leads to higher levels of PAI-1. COVID-19 risk factors are consistent with this scenario, as PAI-1 levels are increased in hypertension, obesity, diabetes, cardiovascular diseases, and old age. We discuss the possibility of using various approved drugs, or drugs currently in clinical development, to treat COVID-19. This perspective suggests to enhance STAT1 activity and/or inhibit STAT3 functions for COVID-19 treatment. This might derail the escalating STAT3/PAI-1 cycle central to COVID-19.
Source:
 mix.com
mix.com
You've added this content to your favorites.

Post your comment
Load More
